Introduction
The use of antifungal drugs in human and veterinary medicine has increased in recent years. In humans, increased susceptibility to fungal infections has arisen because of immunosuppression secondary to HIV infection or treatment with potent immunosuppressive drugs. In animals, opportunistic fungal infections also result from immunosuppression, and the importance of fungal infections in dogs and cats is increasingly recognized. Antifungal drugs used in dogs and cats are generally not approved for use in these species. Therefore, veterinarians use human antifungal drugs off-label, with indications and dosing protocols often extrapolated from the human use. Because of differences in pharmacokinetics among species for these drugs, and differences in susceptibility to adverse effects in dogs and cats compared to people, this approach is not optimal. Fortunately, the clinical experience and pharmacokinetic data have expanded for these human drugs, which will improve antifungal treatment in animals. Efforts also have focused on the development of new, less toxic, and more efficacious antifungal drugs. This chapter reviews the classification, mechanisms of action, spectrum of activity, resistance mechanisms, tissue penetration, clinical use, and adverse effects of the major systemic antifungal drugs used to treat dogs and cats.
Antifungal drug treatment is often more prolonged than antibacterial drug treatment. One reason for this difference is that fungal organisms grow more slowly, and the drugs used (primarily azoles) are fungistatic, not fungicidal. Therefore, long-term treatment is needed to inhibit fungal growth and allow for the animal’s immune system, which is often compromised, to eradicate the infection. Treatment of animals with deep mycoses with antifungal drugs must often be continued for months, and in some animals it may be weeks before clinical improvement is evident. Sometimes, disease worsens in the first week of treatment, because of the host’s inflammatory response to killed organisms. This may have severe consequences for the host when there is extensive involvement of the pulmonary parenchyma or the central nervous system (CNS). Concurrent treatment with a non-steroidal anti-inflammatory drug (NSAIDs) or, when CNS involvement is present, judicious use of a short course of anti-inflammatory glucocorticoids may improve outcome in these situations.
The prognosis for animals with disseminated mold infections that have reversible underlying immunosuppression (such as drug-induced immunosuppression) may be better than the prognosis for animals with no apparent underlying immunosuppressive disease. This is because the latter group of animals are likely to have underlying (irreversible) genetic defects in immunity.
Azole Antifungals
Classification:
Azole antifungals are classified as imidazoles or triazoles based on whether they possess two or three nitrogen molecules in their azole ring. Ketoconazole, enilconazole, and clotrimazole are imidazoles. The latter two drugs have poor oral bioavailability and are used topically in veterinary medicine for the treatment of superficial mycoses. Triazole antifungal drugs, such as itraconazole and fluconazole, are more slowly metabolized and have less impact on mammalian sterol synthesis than do imidazoles. Imidazoles and triazoles have been widely used to treat a variety of mycoses, which include candidiasis, cryptococcosis, blastomycosis, histoplasmosis, coccidioidomycosis, dermatophytosis, sporotrichosis, and aspergillosis.
Mechanism of Action
Azole antifungal drugs inhibit sterol 14α-demethylase, a cytochrome P450–dependent fungal enzyme involved in synthesis of ergosterol, a key component of the fungal cell wall, from lanosterol. The result is the accumulation of 14α-methylsterols, which disrupt the fungal cell membrane.
The majority of the adverse effects and drug interactions observed with these drugs relate to the cross-inhibition of mammalian P450 enzymes.
All of the azole antifungals have the potential to be teratogenic, and their use should be avoided in pregnancy.
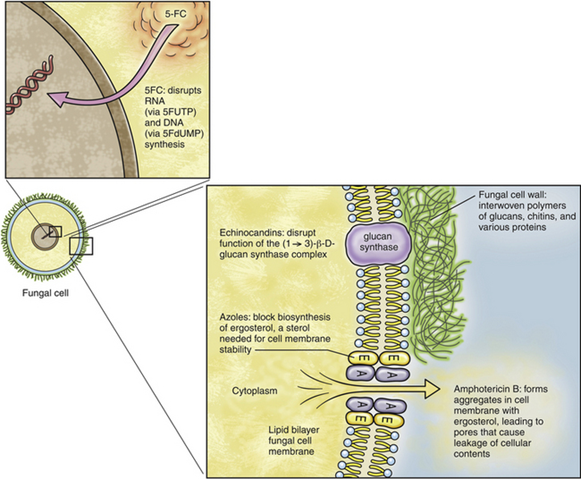
Mechanism of action of antifungal drugs. 5FC, 5-flucytosine; 5FUTP, 5-fluorouridine triphosphate; 5FdUMP, 5-fluorodeoxyuridine monophosphate; A, amphotericin B; E, ergosterol.
Spectrum of Activity
Spectrum of Activity and Tissue Distribution of Different Antifungal Drugs
| Drug | Spectrum of Activity | Tissue Distribution |
| Ketoconazole | Dimorphic fungi, Malassezia. Ineffective for aspergillosis | Skin, bone, joint, lung. Poor CNS penetration |
| Itraconazole | Dimorphic fungi and molds, Malassezia spp. | Skin, bone, lung. May enter the CNS and eye with inflammation |
| Fluconazole | Some Candida spp., Malassezia spp., some dimorphic fungi. Poor activity against molds. Aspergillus spp. are intrinsically resistant | Widely distributed, including to the skin, lung, CNS, urine, and eye |
| Voriconazole | Dimorphic fungi, yeasts, and molds with the exception of Sporothrix schenckii and zygomycetes | CNS, eye, lung, bone |
| Posaconazole | Dimorphic fungi, yeasts, and molds including zygomycetes | Widely distributed |
| Amphotericin B | Broad spectrum. Also active against Leishmania | Limited penetration of the CNS and eye |
| 5-Flucytosine | Cryptococcus and Candida spp. | Widely distributed, which includes the CNS, eye, and urine |
| Griseofulvin | Dermatophytes | Concentrates in skin |
| Terbinafine | Activity highest for dermatophytes. To a lesser extent may have activity against other dimorphic and filamentous fungi | Concentrates in skin and hair |
| Caspofungin | Candida and Aspergillus spp. Not active against Cryptococcus spp. or when given alone to treat Coccidioides | Widely distributed. Poor penetration of the CNS and eye |
.
Suggested Azole Antifungal Drug Doses for Dogs and Cats

C, Cats; D, dogs
Resistance:
Unfortunately, resistance to azole antifungal drugs has emerged among some fungi. Resistance results from mutations in the gene encoding the demethylase enzyme, increased production of C-14α demethylase, and increased azole efflux by fungal cell membrane transporters. Methods for in vitro susceptibility testing have become more standardized, with improved correlation between the results of in vitro susceptibility testing and clinical response, but breakpoints (criteria that define susceptible versus resistant minimum inhibitory concentration values) are still needed for many drug-fungus combinations. Increasingly, it is apparent that resistance to one azole does not always imply resistance to other azole antifungal drugs.
Ketoconazole
Spectrum of Activity and Clinical Use
The use of ketoconazole has largely been replaced by itraconazole for treatment of many mycoses, because of the greater toxicity and reduced efficacy of ketoconazole when compared with triazole antifungal drugs. Because of its low cost, ketoconazole continues to be used in veterinary medicine when the cost of other antifungal drugs is prohibitive to the client, and it remains efficacious for treatment of Malassezia dermatitis and feline nasal and cutaneous cryptococcosis. The absorption of ketoconazole is improved when it is administered with food, but it is inhibited by concurrent use of antacids. Ketoconazole is highly protein bound and is metabolized extensively by the liver. Nevertheless, moderate hepatic dysfunction does not alter blood levels of ketoconazole. Inactive products are excreted in bile and, to a lesser extent, the urine. Because of poor CNS penetration, it is ineffective for treatment of meningeal cryptococcosis and aspergillosis.
Adverse Effects
The most common adverse effects of ketoconazole in dogs and cats are vomiting, anorexia, lethargy, and diarrhea. Administration of ketoconazole with food may reduce gastrointestinal adverse effects. Mild increases in the activity of serum transaminases occur commonly during treatment, but do not warrant discontinuation of the drug. Less commonly, ketoconazole causes hepatitis, which may be accompanied by anorexia, vomiting, lethargy, increasing activities of serum ALT and ALP, and hyperbilirubinemia. The drug should be discontinued if this occurs and serum chemistry values checked 1 to 2 days later. Hepatitis can occur at any time, and the onset may be extremely rapid. Pruritis and cutaneous erythema have been reported in fewer than 1% of dogs treated with ketoconazole. Lightening of the hair coat color and cataract formation occur rarely.
Ketoconazole is a potent inhibitor of mammalian cytochrome P450 enzymes and efflux transporter proteins such as P-glycoprotein. It also inhibits testosterone and cortisol synthesis. As a result, it has been used to treat pituitary-dependent hyperadrenocorticism in dogs and also to deliberately inhibit the metabolism of cyclosporin, allowing a reduction in dose and, therefore, cost, although there are concerns about whether use of ketoconazole in this way could contribute antifungal drug resistance and it increases the risk of adverse drug reactions. Transient infertility can occur during treatment of intact male animals. Ketoconazole can interfere with P-glycoprotein transport of ivermectin, which predisposes dogs to ivermectin toxicity.
Itraconazole
Spectrum of Activity and Clinical Use
Itraconazole is one of the most widely used azoles in veterinary medicine. It has been used to treat blastomycosis, sporotrichosis, aspergillosis, coccidioidomycosis, dermatophytosis, histoplasmosis, phaeohyphomycosis, paecilomycosis, cryptococcosis, and Malassezia infections. It is available in capsules (which contain itraconazole granules) and an oral solution. The IV solution has been withdrawn. Itraconazole in capsules is best absorbed when given with food because the acid secretion stimulated with feeding increases the drug solubility, which is necessary for dissolution and absorption. However, the oral solution is complexed with cyclodextrin to improve solubility, and administration of food does not influence absorption of this formulation. Absorption of the oral solution is consistently better than absorption of the capsule formulation, regardless of feeding. As with ketoconazole, concurrent administration of gastric acid suppressants (H2-blockers, proton pump inhibitors) may reduce absorption of the oral capsule. In cats, the oral solution is very well absorbed, and dose reduction is indicated to reduce the chance of toxicity. In addition to hydroxypropyl-β-cyclodextrin, this formulation contains sorbitol, propylene glycol, hydrochloric acid, cherry flavor, and saccharin, which may be unpalatable for cats. In Europe and Canada an oral solution for cats is available (Itrafungol, 10 mg/mL). Compounded formulations prepared by pharmacists have highly variable, poor, and even negligible oral absorption in animals, in addition to being unstable formulations. As a result, compounded formulations should never be used.
Like ketoconazole, itraconazole undergoes hepatic metabolism and inhibits metabolism of other P450-dependent drugs (e.g., cisapride, diazepam, cyclosporin). It is the only triazole antifungal drug that is converted to an active metabolite, hydroxylitraconazole. Itraconazole is highly (99%) plasma protein bound and does not appear in urine, cerebrospinal fluid (CSF), or ocular tissues, although penetration of the CSF and eye can occur in the presence of inflammation. Itraconazole accumulates in the skin and claws and is the drug of choice for treatment of dermatophytosis in cats. Itraconazole is also a good choice for treatment of fungal osteomyelitis. Although loading doses have been recommended, these initial high doses can increase the risk of toxicity and do not appear to improve outcome. Advanced liver disease increases itraconazole concentrations. Because of the variable absorption of oral itraconazole, monitoring of steady-state plasma concentrations (e.g., 3 weeks after initiating treatment) may be helpful if clinical responses are suboptimal. Trough plasma itraconazole concentrations should be 0.5 to 1 µg/mL as determined using high-performance liquid chromatography.
Adverse Effects
The most common adverse effects of itraconazole in dogs and cats are vomiting and anorexia. Division of the total dose for twice-daily administration has been associated with decreased gastrointestinal signs and improved absorption in human patients. Gastrointestinal adverse effects may be more common with the oral solution. Mild to moderate increases in serum ALT activity commonly occur during treatment. Provided these are not accompanied by inappetence, treatment need not be discontinued. Significant hepatotoxicity, accompanied by anorexia, vomiting, and hyperbilirubinemia, is less likely to occur than with ketoconazole but has been reported in humans as well as cats and dogs. Hepatotoxicity more commonly occurs in dogs treated with 10 mg/kg/day. Ulcerative skin lesions can occur in dogs, especially when doses of 10 mg/kg/day or higher are used. Development of hepatitis or severe cutaneous ulceration should prompt discontinuation of the drug. It may be possible to reinstitute treatment at a lower dose once the adverse effects have resolved. Occasionally mild, focal cutaneous ulcerative lesions can resolve spontaneously, without discontinuation of treatment.

Submandibular ulcerative skin lesions in a golden retriever that had been treated with itraconazole for systemic blastomycosis.
Itraconazole does not suppress adrenal and testicular function like ketoconazole, but can inhibit the metabolism of other P450 enzyme-dependent drugs, including cyclosporine, digoxin, cisapride, and vinca alkaloids. It can also interfere with P-glycoprotein transport of ivermectin, which results in increased plasma ivermectin concentrations.
Fluconazole
Spectrum of Activity and Clinical Use
Fluconazole is the least active azole antifungal drug and has the narrowest spectrum. The activity of fluconazole is limited to some Candida spp., Cryptococcus spp., Malassezia spp., and some dimorphic fungi. Fluconazole has poor activity against molds, and Aspergillus species are intrinsically resistant to fluconazole. Some Cryptococcus isolates are resistant to fluconazole, and resistance can develop during treatment. Itraconazole is preferable to fluconazole for treatment of histoplasmosis, blastomycosis, sporotrichosis, and dermatophytosis, although fluconazole has been successfully used to treat coccidioidomycosis, blastomycosis, and Malassezia infections in dogs.
Fluconazole is available as tablets, an oral suspension, and as an IV solution. The availability of generic fluconazole has greatly reduced its cost and increased its use in veterinary medicine. Approved generic fluconazole tablets are bioequivalent to the brand name formulation in people. Similar bioequivalence is likely in animals. Fluconazole is more water-soluble and stable than itraconazole, and the compounded oral suspensions can be used in animals with a 14-day beyond-use-day (BUD). Some compounded preparations may have reduced activity. Fluconazole is almost completely absorbed after oral administration, and bioavailability is not altered by food or gastric acidity. In contrast to itraconazole and ketoconazole, fluconazole is only weakly protein bound. Because it is more water soluble than other azoles, fluconazole diffuses into body fluids such as saliva, urine, synovial fluid, and CSF. Therefore, it is the drug of choice for treatment of susceptible meningeal and urinary tract fungal infections. Renal excretion accounts for more than 90% of the elimination of fluconazole. The dosage should be decreased in animals with renal failure; this is especially important in patients receiving other P450-dependent drugs.